第十章 电解与库仑分析法
 知识点一:电解分析法
知识点一:电解分析法
一.电解分析的基本原理
(一)电解现象
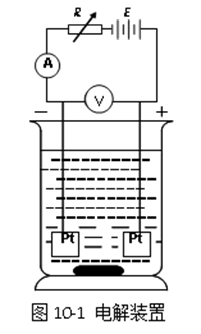
电解是指直流电流通过某种电解质溶液时,在电流的作用下电极溶液界面发生电极反应,从而引起溶液中某种物质分解的过程。例如在硫酸铜溶液中,浸入两个铂电极,电极通过导线分别与直流电源的正极和负极相连接(图10-1)。如果在两电极间有足够大的电压,则可观察到有明显的电极反应:
阴极反应: ![]()
阳极反应: ![]()
于是阳极上有氧气放出,阴极上有金属铜析出,形成金属镀层。
(二)分解电压和析出电位
在电解时,能够使被电解物质在两电极上产生迅速、连续的反应,所需的最低外加电压称为分解电压。当以很小的电压加于电解池,起初没有电流通过。调节电阻R使外加电压增加,则电流略有增加。当达到某一定电压值时,如图10-2中D点,通过电解池的电流有明显的增加,同时在两电极上产生连续不断的电极反应,以后电流随电压的增加而直线上升。D点的电压就是分解电压。一种物质的分解电压对于可逆过程来说,在数值上等于它本身所构成的自发电池的电动势。在电解池中,此电动势称为反电动势。反电动势的方向与外加电压的方向相反,它阻止电解反应的进行。
例如,电解硫酸铜溶液,开始时并不构成自发电池。当接上外电压后,如外电压很小,在最初瞬间会有及少量的Cu和O2分别在阴极和阳极上析出。当一旦有少量的Cu和O2被吸附在电极上时,就立刻形成由氧电极和铜电极组成的自发电池,产生反电动势,阻止电解进行。只有当外加电压达到能克服此反电动势时,电解才能开始进行,电流才能显著上升。所以,要使某一电解过程能够进行,只有当外加电压超过(即使是很微小的数值)它自身构成的自发电池的电动势,也就是它的分解电压时,电解在理论上才成为可能。如以i表示电解电流,R表示电解池内阻,E表示分解电压,Ud表示理论分解电压,则外加电压U与Ud有如下关系:
![]() (10-1)
(10-1)
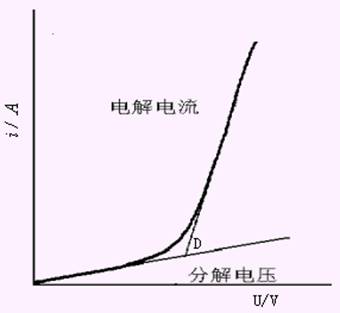
图10-2 电解铜(II)溶液的i-U曲线
在电解过程中,往往只考虑某一电极的电位,即析出电位。析出电位是指物质在阴极是还原析出时所需最正的阴极电位,或阳极氧化析出时所需最负的阳极电位。对于可逆电极反应来说,某物质的析出电位就等于电极的平衡电位。
在上例中,铜电极为阴极,氧电极为阳极,其阴极平衡电位Ec和阳极平衡电位Ea分别为:
![]()
![]()
若铜和氧气原电池的氧分压PO2为21278 Pa,则
![]()
因此,电解硫酸铜溶液时,理论分解电压为:
![]() (10-2)
(10-2)
![]()
如果电解池内阻为0.5 Ω,电解电流i为0.1 A,实际需要的分解电压高达1.68 V,大于理论计算值。这种实际电极电位偏离平衡电位的现象,称为极化现象或极化作用。极化作用的结果产生了过电位。过电位使阳极更正,阴极更负。电解池的过电压η等于阳极过电位ηa和阴极过电位ηc绝对值之和。
有两种极化作用产生过电位:
1.电化学极化产生过电位 若两电极的电流密度大,Mn+离子在阴极上来不及还原,而使阴极表面积累了过量的电子,因而电极电位向负移动。析出金属时,过电位一般很小,可以忽略;而析出气体(如H2,O2)时,过电位很大,必须考虑。
2.浓差极化引起的过电位 在电极表面,Mn+还原速率很大,使电极表面Mn+浓度很快下降。若溶液中Mn+离子向阴极表面的扩散速率小于电极反应速率,则电极表面的Mn+浓度小于溶液中的Mn+浓度,使阴极平衡电位更负些。搅拌可以消除浓差极化。
由于存在极化作用,分解电压的理论方程式应为:
![]() (10-3)
(10-3)
将式(10-3)代入式(10-1)中得
![]() (10-4)
(10-4)
式(10-4)称为电解方程式,它表明实际分解电压是理论分解电压、电池的过电位、电解池中的电压降iR之和。
在上述的电解硫酸铜溶液例子中,阳极过电位ηa是0.72 V,铂电极的过电位ηc忽略不计,电解时外加电压为![]()
二.电解分析法的应用
电解分析方法有两种,一是在电解电流保持恒定的情况下进行电解,称为控制电流电解或恒电流电解;二是控制工作电极的电位为一定数值或在一定电位范围内的电解,称为控制电位电解。
(一)控制电流电解法
1.基本装置
控制电流电解法是在电解过程中不断调节外加电压,使通过电解池的电流恒定。其装置如图10-3所示。以串联蓄电池作为直流电源,电源通过可调电阻R与两电极相连。工作电极为网状铂电极,具有较大的表面积。在允许的电流密度下可以使用较大的电解电流,以加快电解速度。另外,网状铂电极也有利于溶液的搅动,可以减小浓差极化。辅助电极是螺旋状或平板状铂阳极,电解时兼作搅拌器。通过电解池的电流一般为0.5~5 A。
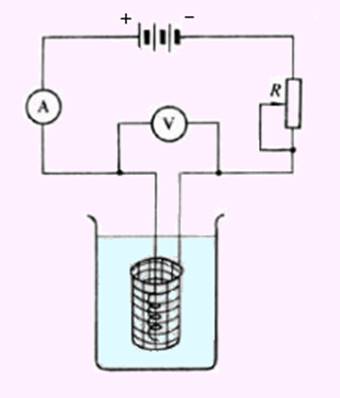
图10-3 控制电流电解装置
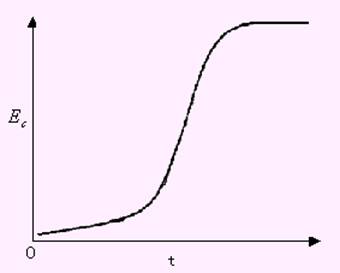
图10-4 控制电流电解的E-t曲线
2.阴极电位随时间变化曲线
在控制电流电解分析中,阴极电位Ec与电解时间t的关系曲线如图10-4所示。由图可知,随着电解的进行,阴极表面附近Mn+浓度不断降低,电极电位变负。经过一段时间后,因Mn+浓度较低,使得阴极电位改变的速率变慢,Ec-t曲线上出现较为平坦部分。与此同时,电极电流也不断降低,为了维持电极电流恒定,就必须增大外加电压,使阴极电位更负。这样,由于静电引力作用使Mn+以足够快的速度迁移到阴极表面,并继续发生电极反应以维持电流恒定,Mn+继续在阴极上还原析出,直到电解完全,这就是控制电流电解原理。
3.特点和应用
恒电流电解不控制阴极电位,靠不断增大外加电压保持一个较大的、基本恒定的电解电流,因而电解效率高,分析速度快。但是,由于在电解过程中不控制阴极电位,随着电解的进行,阴极电位逐渐变负,若溶液中有几种离子共存时,在还原电位较正的离子还未完全析出时,阴极电位可能已负到另一种离子的析出电位而使其伴随析出。所以,这种方法的选择性较差。然而,恒电流电解法可以有效地使一种还原电位正于氢的元素与还原电位负于氢的各种元素分离,进而准确地加以测定。常用恒电流电解法测定的元素见表10-1。
表10-1 常用控制电流电解法测定的元素
测 定 离 子 |
称 量 形 式 |
条 件 |
Cd2+ |
Cd |
碱性氰化物溶液 |
(二)控制电位电解法
控制电位电解法包括控制阴极电位电解分析法和控制阳极电位电解分析法两种。其中最重要的是控制阴极电位电解分析法。
1.基本装置
控制阴极电位电解过程需要随时测量阴极电位并随时调节外加电压以控制电极电位为一恒定值。基本装置如图10-5所示。
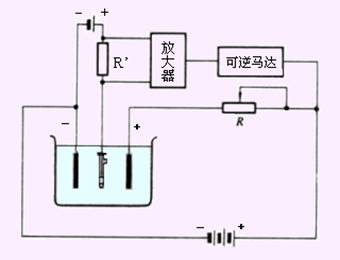
图10-5 控制阴极电位电解装置
由甘汞电极、铂网电极和电位计组成控制阴极电位系统。电位计可显示相对于甘汞电极电位的阴极电位数值。由直流电源、可变电阻R及电解池组成电解装置。电解时,通过不断调节可变电阻R以调节外加电压的大小,进而调节阴极电位。
2.阴极电位的选择
溶液中存在两种以上可沉积的离子时,进行电解就应考虑干扰和分离问题。如果两种金属离子的还原电位相差较大,可用控制阴极电位电解法使两种金属分离。例如,溶液中有A、B两种金属离子,其电解电流与阴极电位i-Ec曲线如图10-6 所示。图中a、b两点分别为A、B两种金属离子的析出电位。只要将阴极电位控制在ab之间进行电解,如图中d点,就可以使A离子定量析出,而B离子仍然保留在溶液中,从而实现A、B离子的分离和A离子的测定。待A离子测定完毕后,再将阴极电位控制在c点进行电解,就可以实现对B离子的测定。
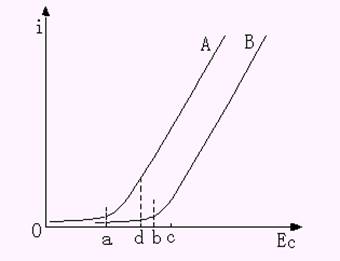
图10-6 分离A、B两种离子的i-Ec曲线
从Nernst方程可见,要使发生电极反应的离子浓度减小10倍,阴极电位只须负移 V。可以计算出,离子浓度降低105倍,阴极电位负移 V。一般认为,被测离子浓度降到110-6 mol/L或110-7 mol/L时,可认为达到分离和分析的要求。因此,起始浓度大致相同的两种一价离子,只要其标准电位相差0.3 V以上,就可以控制阴极电位电解使其定量分离。对于二、三价的离子,所要求的标准电位相差分别为0.15 V和0.10 V。
应用控制阴极电位电解法对铜、铋、铅、锡四种共存离子进行分离和测定就是一个很好的例子。在中性的酒石酸盐溶液中,阴极电位为-0.2 V(相对于甘汞电极)时,铜首先析出。经称量后,再将镀了铜的电极放回溶液,在-0.4 V电位下电解,使铋定量析出。再将阴极电位调节到-0.6 V电解,此时铅定量析出。然后,酸化溶液,使锡的酒石酸络合物分解,在-0.65 V阴极电位下电解,锡就定量沉积下来。显然,只要阴极电位控制得适当,就能很容易地定量测定多种离子化合物中的某一种离子。
3.电流-时间曲线
在控制电位电解过程中,由于被测离子在阴极上不断析出,电流随着时间增长不断减小,在某金属离子完全析出后,电流应降至零。但由于残余电流的存在,电流最后达到恒定的背景电流值,见图10-7。对于仅有一种离子在电极上还原,电流效率为100%时,电流和时间的关系为:
![]() (10-7)
(10-7)
式中it为t时的瞬时电流;i0为初始电流;k为常数,它与电极表面积、溶液体积、搅拌速度以及电极反应类别等因素有关。
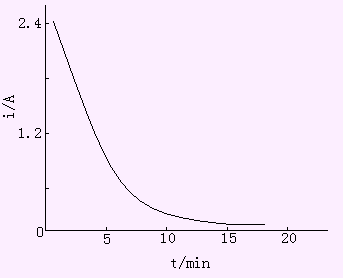
图10-7 控制阴极电位电解的i-t曲线
4.特点及应用
由于控制阴极电位能有效地防止共存离子的干扰,因此选择性好。该法既可作定量测定,又可广泛地用作分离技术,常用于多种金属离子共存情况下某一种离子含量的测定。这种方法的一些应用实例列于表10-2。
表10-2 控制电位电解法应用实例
测 定 元 素 |
分离的干扰元素 |
Ag |
Cu和碱金属 |
请同学们继续学习
