第五章 红外光谱法
 知识点二:红外光谱与分子结构
知识点二:红外光谱与分子结构
红外光谱的最大特点是具有特征性,这种特性与各种化学键振动的特征相联系。大多数有机化合物是由C、H、O、N、S、P卤素等元素组成,而其中最主要的是C、H、O、N四种元素。因此可以说大部分有机化合物的红外光谱基本上是由这四种元素所形成的化学键的振动贡献的。按照光谱特征与分子结构的关系,红外光谱可以分为基频区和指纹区两大区域。
一、红外光区的划分
红外光谱区在可见光区和微波光区之间,波长范围为0.75~1000 μm。根据实验技术和应用的不同,通常将红外区划分成三个区(表5-1):近红外光区(0.78~2.5μm ),中红外光区(2.5~25μm )和远红外光区(25~1000μm)。
其中中红外区是研究和应用最多的区域,一般说的红外光谱就是指中红外区的红外光谱。
表5-1 红外光谱区划分
红外吸收光谱一般用T-曲线或T-σ曲线来表示。纵坐标为百分透射比T%因而吸收峰向下,向上则为谷;横坐标是波长或波数σ(单位为cm-1)。
红外光谱的最大特点是具有特征性,这种特征性与各种化学键振动的特征相联系。大多数有机化合物是由C、H、O、N、S、P卤素等元素组成,而其中最主要的是C、H、O、N四种元素。因此可以说大部分有机化合物的红外光谱基本上是由这四种元素所形成的化学键的振动贡献的。按照光谱特征与分子结构的关系,红外光谱可以分为基频区和指纹区两大区域。
1、基频区(4000-1350 cm-1)
基频区又称特征区或官能团区。已经发现具有某些官能团或结构基团的化合物的振动频率几乎与分子的其余部分无关,这些频率是有关功能团和结构基团的特征,因此被称为基团频率。对多数有意义的基团来说,其特征伸缩振动频率应该在4000 - 1500 cm-1范围内。所以,4000-1350 cm-1 区常被称为基频率区。基团频率区又可分为三个区域:
(1)X-H伸缩振动区 (4000-2500 cm-1)
X可以是O、H、C或S原子。O-H的伸缩振动在3700~3100 cm-1,醇、酚、有机酸和水分子在此区域有较强的吸收。N—H伸缩振动在3500~3300 cm-1,伯、仲酰胺和伯、仲胺类在此区域内都有吸收谱带,N—H谱带与O-H谱带重叠,但N—H吸收峰尖锐,O-H谱带常由于氢键的存在,频率降低,谱带变宽。饱和烃C-H伸缩振动在3000 cm-1以下,不饱和烃(包括烯烃、炔烃、芳烃)C-H伸缩振动在3000 cm-1以上。
(2)叁键和累积双键区 (2500-1900 cm-1)
主要包括-C≡C、-C≡N等叁键的伸缩振动,以及-C=C= C、-C=C=O等累积双键的不对称伸缩振动。
(3)双键伸缩振动区 (1900-1200 cm-1)
该区域主要包括C=O、C=C、C=N、N=O等的伸缩振动和苯环的骨架振动,以及芳香族化合物的倍频谱带。
-C=O的伸缩振动出现在1850-1600 cm-1,是红外谱图中最强的吸收峰,C= O伸缩振动的吸收峰是判断有无羟基存在的主要依据。
-C=C的伸缩振动出现在1680-1620 cm-1,它的强度一般很弱。单环芳烃的- C= C伸缩振动(即芳环的骨架振动)在1620~1450 cm-1范围内有四个吸收峰,中1480 cm-1和1620~1590 cm-1区域的两个吸收峰是判断芳环是否存在的重要标志。
苯的衍生物在2000-1667 cm-1区域出现C-H面外弯曲振动的泛频峰。虽然强度很弱,但吸收峰形状和数目与芳环的取代类型有关。利用该区的吸收峰与900~600 cm-1区域苯环的C-H面外弯曲振动吸收峰可以确定苯环的取代类型。图5-3给出了几种不同的苯环取代类型在这两个区域的吸收峰图形:
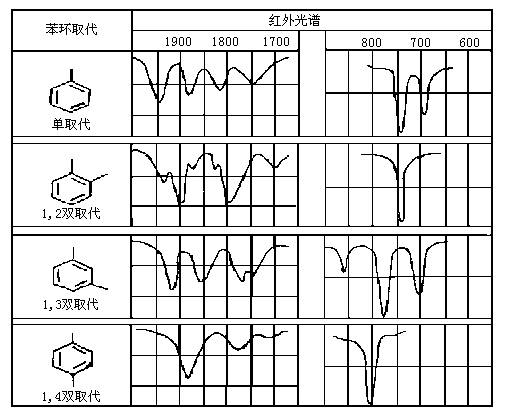
图5-3苯环取代类型对红外光谱的影响
(4)X-H弯曲振动区(1650~1350 cm-1)
这个区域包括出-H、N-H弯曲振动。甲基在1380~1370 cm-1出现一个很特征的弯曲振动吸收峰,这个吸收峰的位置很少受到取代基的影响,干扰也较少,可作为判断甲基是否存在的依据。当一个碳原子上存在两个甲基时,两个甲基的弯曲振动互相耦合而使1370 cm-1附近的吸收峰发生分裂,出现两个吸收峰 。
2、指纹区(1350-650 cm-1)
指纹区的吸收峰是由于C-C、C-O、C-X单键的伸缩振动以及分子骨架中多数基团的弯曲振动所引起的。由于各种单键的强度大体相同,相邻单键之间的相互作用,使得这个区域内的吸收光谱变得非常复杂,并且对结构上的微小变化非常敏感,只要在化学结构上存在细小的差异(如同系物、同分异构体和空间构象等),在指纹区就明显的有反映。犹如人的指纹一样,两个人的指纹不可能完全相同,两个化合物的红外光谱在指纹区也不相同。因此,指纹区对鉴别化合物是很有用的。
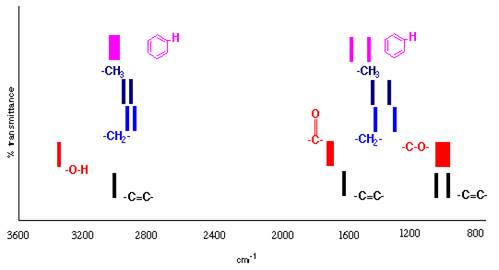
图5-4 给出了部分官能团红外吸收频率的大致位置。
图5-4红外特征吸收频率的大致位置
指纹区可以分为两个区域:
1.1300-900 cm1区域是C-O、C-N、C-F、C-P、C-S、P-O、Si-O等单键的伸缩振动和C=S、S=O、P=O等双键的伸缩振动吸收。
2.900-650 cm1区域内的某些吸收峰可用来确认化合物的顺反构型。
表5-2对一些基团的红外吸收谱带进行了简要的总结。
表5-2 红外光谱中一些基团的吸收频率
区 域 |
基 团 |
吸收频率/ cm-1 |
振动形式 |
吸收强度 |
说 明 |
|
基频区 |
XH伸缩振动区 |
—OH(游离) |
3650~3580 |
伸缩 |
m,sh |
判断有无醇类、酚类和有机酸的重要依据
末端=C—H在3085 cm-1附近 三元环中的 |
—OH(缔合) |
3400~3200 |
伸缩 |
s,b |
|||
—NH2,—NH(游离) |
3500~3300 |
伸缩 |
m |
|||
—NH2,—NH(缔合) |
3400~3100 |
伸缩 |
s,b |
|||
—SH |
2600~2500 |
伸缩 |
|
|||
不饱和C—H |
3000以上 |
伸缩 |
|
|||
≡C—H |
3300附近 |
伸缩 |
s |
|||
=C—H |
3010~3040 |
伸缩 |
s |
|||
苯环中的C—H |
3030附近 |
伸缩 |
s |
|||
饱和C—H |
3000~2800 |
伸缩 |
|
|||
—CH3 |
2960±5 |
不对称伸缩 |
s |
|||
—CH3 |
2870±10 |
对称伸缩 |
s |
|||
—CH2 |
2930±5 |
不对称伸缩 |
s |
|||
—CH3 |
2850±10 |
对称伸缩 |
s |
|||
区 域 |
基 团 |
吸收频率/ cm-1 |
振动形式 |
吸收强度 |
说 明 |
|
基频区 |
三键区 |
—C≡H |
2260~2220 |
伸缩 |
s |
针状,干扰少 R—C≡C—H在2100~2140 cm-1;R’—C≡C—R在2190~2260 cm-1;若R’=R,对称分子,无红外谱带 |
—N≡N |
2310~2135 |
伸缩 |
m |
|||
—C≡C— |
2600~2100 |
伸缩 |
v |
|||
=C=C— |
1950附近 |
伸缩 |
v |
苯环的骨架振动
其它吸收谱带干扰少,是判断羰基(酮、酸、酯、酸酐等)的特征频率,位置变动大 |
||
双键伸缩振动区 |
C=C |
1680~1620 |
伸缩 |
m,v |
||
芳环中C=C |
1600,1580 |
伸缩 |
v |
|||
1500,1450 |
||||||
—C=O |
1850~1600 |
伸缩 |
s |
|||
—NO2 |
1600~1500 |
不对称伸缩 |
s |
|||
—NO2 |
1300~1250 |
不对称伸缩 |
s |
|||
S=O |
1220~1040 |
伸缩 |
s |
|||
XH弯曲振动区 |
—CH3,—CH2 |
1460±10 |
CH3不对称弯曲 |
m m |
大部分有机化合物都含CH3、CH2基,故此峰经常出现
烷烃中CH3基的特征吸收 |
|
—CH3 |
1380~1370 |
对称弯曲 |
m |
|||
—NH2 |
1650~1560 |
弯曲 |
m~s |
|||
指纹区 |
C—O |
1300~1000 |
伸缩 |
s |
C—O键(酯、醚、醇)的极性很强,故强度大,常成为谱图中最强的吸收。
n>4 |
|
C—O—C |
1150~900 |
伸缩 |
s |
|||
C—F |
1400~1000 |
伸缩 |
s |
|||
C—Cl |
800~600 |
伸缩 |
s |
|||
C—Br |
800~600 |
伸缩 |
s |
|||
C—I |
500~200 |
伸缩 |
s |
|||
=CH2 |
910~890 |
面外摇摆 |
s |
|||
—(CH2)n— |
720 |
面内摇摆 |
v |
|||
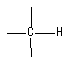 出现在2890 cm-1,很弱
出现在2890 cm-1,很弱
二、影响基团频率的主要因素
分子中各基团的振动并不是孤立的,要受到分子中其他部分,特别是临近基团的影响,有时还会受到溶剂、测定条件等外部因素的影响。因此,同一基团的振动在不同结构中或不同环境中其吸收频率总会有些差异。引起基团频率位移的因素大致分为两类,即内部结构因素和外部环境因素。
1、内部因素
(1)诱导效应
由于邻近基团具有不同的电负性,通过静电诱导效应引起基团中电荷分布的变化,从而改变了键的力常数,使键或基团的特征频率发生位移。
例如,一般电负性大的基团或原子吸电子能力强,与烷基酮羰基上的碳原子数相连时,由于诱导效应就会发生电子云由氧原子转向双键的中间,增加了C=O键的力常数,使C=O的振动频率升高,吸收峰向高波数移动。随着取代原子电负性的增大或取代数目的增加,诱导效应越强,吸收峰向高波数移动的程度越显著。
(2)共轭效应
包括π-π、p-π共轭,由于轨道耦合,使共轭体系中的电子云密度平均化,结果使双键电子云密度降低,力常数减小,吸收峰向低波数方向移动。在一个化合物中诱导效应和共轭效应经常同时存在,此时,吸收峰的移动方向取决于哪一种效应占优势。
(3)氢键
形成氢键后,X-H的振动频率降低,吸收向低波数移动,吸收强度增大,谱带变宽。分子间的氢键与浓度和溶剂的性质有关,随浓度减小而消失,而分子内氢键不受溶液浓度的影响,二者易于区别。
(4)振动偶合
当两个振动频率相同或相近的基团相邻具有一公共原子时,由于一个键的振动通过公共原子使另一个键的长度发生改变,产生一个“微扰”,从而形成了强烈的振动相互作用。其结果是使振动频率发生感变化,一个向高频移动,另一个向低频移动,谱带分裂。振动耦合常出现在一些二羰基化合物中,如,羧酸酐。
(5)费米共振
当一种振动的倍频与另一种振动的基频接近时,而发生相互作用,导致谱带分裂,这种现象就叫费米共振,导致原来的弱倍频吸收强度显著增加。
例如,大多数醛在2850~2720 cm-1范围内会出现两个中等强度的吸收,是醛基的C-H伸缩振动与C-H键的弯曲振动(1390 cm-1)的倍频之间发生费米共振所致。
2、外部因素
同一物质的不同状态,由于分子间相互作用力不同,所得到光谱往往不同。分子在气态时,其相互作用力很弱,此时可以观察到伴随振动光谱的转动精细结构。
液态和固态分子间作用力较强,在有极性基团存在时,可能发生分子间的缔合或形成氢键,导致特征吸收带频率、强度和形状有较大的改变。例如,丙酮在气态时的C-H为1742 cm-1,而在液 态时为1718 cm-1。在溶液中测定光谱时,由于溶剂的种类、溶剂的浓度和测定时的温度不同,同一种物质所测得的光谱也不同。通常在极性溶剂中,溶质分子的极性基团的伸缩振动频率随溶剂极性的增加而向低波数方向移动,并且强度增大。因此,在红外光谱测定中,应尽量采用非极性的溶剂。
请同学们继续学习
